Les travaux qui ont été publiés dans la revue Proceedings of the National Academy of Sciences (PNAS) le 6 juin dernier ouvrent la voie à une conception plus sûre et plus efficace de nouveaux médicaments.
L’observation et la compréhension de la liaison protéine-ligand est fondamentale pour la recherche biomédicale : elle permet de calculer l’affinité et le temps d’union de la molécule médicamenteuse avec sa protéine cible, connaître quelles sont leurs interactions et savoir ainsi comment agit le médicament. Seulement ce processus se déroule à des échelles qui rendent les observations difficiles : la taille des liaisons est de l’ordre de l’Angström (10-10 mètres, échelle d’un atome) sur des durées d’environ 100 nanosecondes.
Filmer ou capturer des déplacements si rapides de molécules avec une résolution spatiale si fine est aujourd’hui impossible. Pour contourner les problèmes liés à l’observation directe, les scientifiques ont donc développé des méthodes de simulation numérique qui permettent de représenter et » voir » les molécules et leurs mouvements à l’échelle atomique avec une grande précision. Sous la houlette de Gianni Fabritiis, coordinateur du Laboratoire de Biophysique Informatique et du Programme de Recherche en Informatique Biomédicale (GRIB) entre l’IMIM et l’UPF, ces travaux donnent ainsi l’accès à des processus biochimiques jusqu’ici invisibles et méconnus.
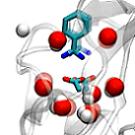
Concrètement, les chercheurs ont reconstruit le processus complet de la liaison d’un inhibiteur enzymatique, le complexe trypsine-benzamidine, grâce à près de 500 simulations moléculaires d’une durée de 100 nanosecondes chacune. Sur la vidéo on peut voir la protéine en arrière plan (en forme de pelote de laine), le complexe trypsine-benzamidine (en vert) et les molécules d’eau (rouge et blanc). Lors de la liaison, la reconnaissance de la molécule par la protéine ciblée provoque une réponse biologique : la compréhension de cette union est donc d’une importance vitale pour la conception de nouveaux médicaments.
À ce jour, c’est la première étude qui fournit une reconstruction complète d’un processus de liaison protéine-ligand. « La méthode nous donne non seulement l’affinité de liaison et la cinétique de la réaction, mais également des informations à l’échelle atomique durant le processus : les sites de liaison, les états de transition et les états métastables, qui sont potentiellement utiles pour augmenter la probabilité de succès dans la conception de médicaments. Cette méthodologie est directement applicable à d’autres systèmes moléculaires, et donc d’intérêt général en matière de recherche biomédicale et pharmaceutique » explique Gianni Fabritiis.
Les chercheurs travaillent maintenant à étendre l’application de cette méthodologie et à optimiser l’usage des capacités de calcul, car dans le cas où les ligands sont plus grands et plus flexibles et où les protéines ont des processus liant plus complexes, un effort de calcul toujours plus grand est exigé.
Source : http://www.bulletins-electroniques.com/actualites/67293.htm